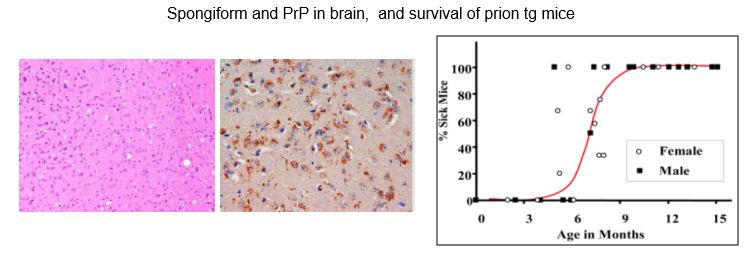
We use our TgMHu2ME199K mice, a model for genetic (E200K) Creutzfeldt-Jakob disease in Libyan Jews. Neurological dysfunction starts at 5 months. Neuronal abnormalities include vacuolation, gliosis, neuronal death, accumulation of the aberrant PrP protein (denominated PrPSc) as well as oxidation of lipids and reduced neuroenesis.
References
Friedman-Levi Y, Mizrahi M, Frid K, Binyamin O, Gabizon R (2013) Prp(ST), a soluble, protease resistant and truncated prp form features in the pathogenesis of a genetic prion disease. PloS one 8(7):e69583
Friedman-Levi Y, Meiner Z, Canello T, Frid K, Kovacs GG, Budka H, Avrahami D, Gabizon R (2011) Fatal prion disease in a mouse model of genetic e200k creutzfeldt-jakob disease. PLoS Pathog 7(11):e1002350
Keller G, Binyamin O, Frid K, Saada A, Gabizon R (2019) Mitochondrial dysfunction in preclinical genetic prion disease: A target for preventive treatment? Neurobiol Dis124:57-66>